Appearance
Superimpose Proteins
The Superimpose Proteins plugin overlays two or more proteins in 3D space for visual comparison and calculates Root Mean Square Deviation (RMSD) values as a numeric metric on structural similarity.
Protein overlay is done either by using backbone (alpha-carbons) or by using all heavy protein atoms; and the alignment can be done on the full protein or limited to a chain.
A Fixed Reference structure is chosen, and Moving Structures are translocated to be superimposed upon the Fixed Reference. RMSD values are reported between the Fixed Reference and each Moving Structure.
Instructions
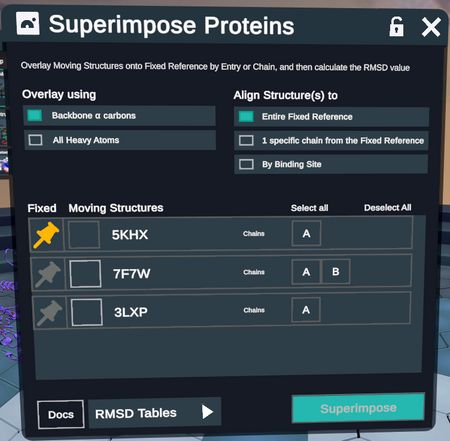
In the upper left, the user can select the Overlay Method.
- Backbone α carbons: Overlay structures based on all paired alpha carbons between the two proteins.
- All Heavy Atoms: Uses backbone atoms and side chain atoms (all protein atoms except for Hydrogen) for paired residues.
In the upper right, there are 4 available alignment modes.
- Align to Entire Fixed Reference: Uses a sequence based alignment to superimpose entire proteins on top of each other.
- 1 specific Chain from the Fixed Reference: Uses a sequence based alignment to superimpose the moving structures onto a specified chain on the Fixed Reference.
- By Binding Site: The user selects a ligand on the Fixed Reference, and the surrounding binding site is aligned to the moving structures using Site-Motif (See Methodologies for more details)
- By Selection: The user selects specific residues in the fixed reference and the moving structures are aligned by that selection only. Users may choose to specify residues on both the fixed and moving structures but only selections in the fixed reference are required for the plugin to run.
A list of proteins in the workspace appears in the main panel. In this panel, choose Fixed Reference and Moving Structures, and subselect a chain or ligand for each, if aligning by chain or binding site, respectively (see 4, below).
By default, the first entry is set as Fixed, and designated by the yellow pin. When selected as Fixed, the box under Moving will be inactive. To change this selection, select the pin next to the desired Fixed Reference protein.
Choose one or more Moving Structures by selecting the boxes under the Moving Structures column. This box will turn green when selected.
Once selections are made, the Superimpose button in the lower right will become active and show the number of proteins selected. (e.g. “Superimpose (3)” for a Fixed Reference and 2 Moving Structures). Click this button to superimpose the proteins and calculate RMSD values. The button will change to “calculating” as the process is being completed.
Once complete, the proteins will be superimposed and locked, for visual inspection. The RMSD tables pulldown menu in the lower left will become active. Each superimpose run will generate a new table, with the most recent run appearing at the top of the pulldown list. Select the run to open the table in the workspace. Tables can be closed and reopened in the workspace.
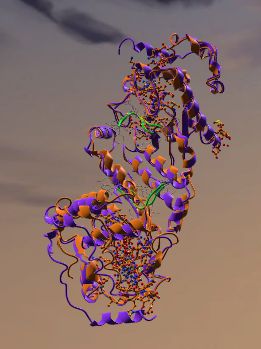
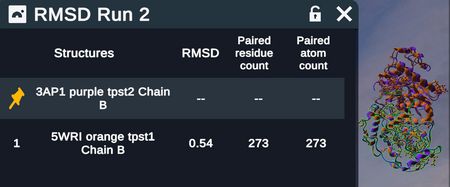
Two superimposed proteins
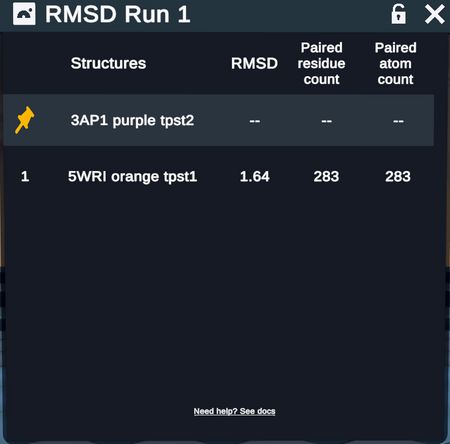
RMSD table describing above superimposed proteins
When aligning by chain, the main panel chain selection boxes will become active. Choose Fixed and Moving Structures as above, and additionally choose a chain for each protein to be superimposed
- Note that selecting a chain on the plugin main panel will also apply green highlighting to the selected chain of the protein structure, to visualize the selections before running.
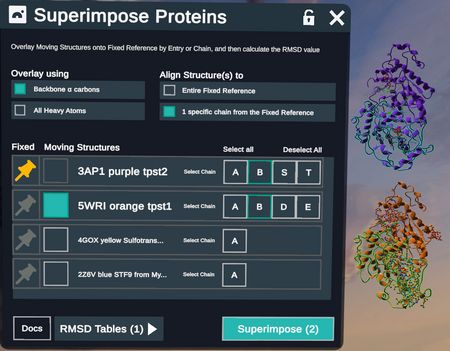
When aligning by Binding Site, the Overlay method will automatically change to All Heavy Atoms.
- An option to Extract binding sites into new entry items appears. This creates a new set of entries containing only the superimposed binding site atoms of each protein.
- Choose Fixed and Moving Structures as above and additionally choose a ligand from the Fixed reference to establish the binding site for superimposition.
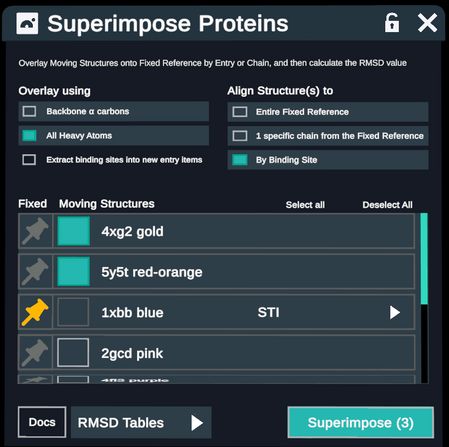
Note the Extract binding sites into new entry items button

When aligning by selection, the user must make a selection in the Fixed Reference. These selections can be made by using the selector tool on the 3D structure itself, or by using menus such as Hierarchy and Antibody Regions table
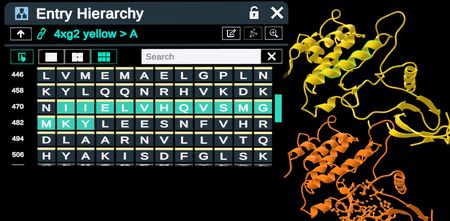
Making a selection in the Fixed Reference using the hierarchy menu.
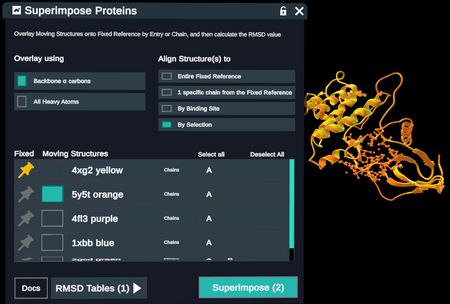
Structures superimposed based on a selection in the fixed reference
RMSD tables report out fixed and moving structures (with chain when relevant) RMSD values, paired residues, and paired atoms
- Note that when superimposing by backbone alpha carbons, the # of paired residues = # of paired atoms; when superimposing by all heavy atoms, the # of paired residues will be ~8x less than the # of paired atoms because there are on average ~8 heavy atoms per residue.
- For two identical proteins, the RMSD would be zero. Values close to zero indicate highly similar proteins. Values over 10 are very different. A combination of visual inspection and RMSD values gives a reasonable indication of how similar the structures are.
Methodologies
We have two primary approaches to superimposing proteins:
Full Protein and Chain alignments both use a sequence based approach.
Binding site overlays use the Site-Motif algorithm, with FPocket for identifying inputs.
Sequence Alignment
Primary modules
Bio.pairwise2
Bio.PDB.Superimposer
Summary
For each pairing of fixed structure and moving structure:
A global alignment is run on protein residue sequences using Bio.pairwise2
Aligned residue atoms are passed into Bio.PDB.Superimposer, which calculates an RMSD value and a transform matrix.
We apply the transform matrix to every moving atom on the complex.
After all moving atoms have been transformed, we update the complexes in the workspace.
Implementation Details
Alignment
- For Entry mode, the entire protein sequence is aligned.
- For Chain mode, only the specified chains are aligned.
Global alignment scoring values
- match = 2
- mismatch = -1
- gap penalty = -10
- gap extend penalty = -.5
Superimpose
- If the alpha carbon overlay method is selected, we only pair the alpha carbons from each residue.
- If all heavy atoms overlay method is selected, we attempt to pair all heavy atoms. In some cases, we cannot get a 1-1 pairing of heavy atoms. In this case, those residues are excluded from the superimpose
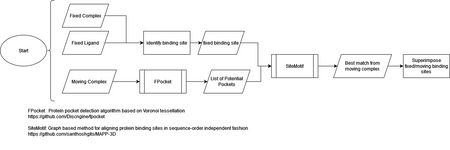
Binding Site Alignments using Site-Motif
Site-motif is described as a graph based method for aligning protein binding sites in sequence-order independent fashion. It can analyze the binding site of a protein and find the best alignment on another protein.
Our algorithm uses FPocket to identify potential binding sites on the moving protein, and those results are compared to the fixed binding site using Site-Motif. Site-Motif calculates alignments of each FPocket result to the fixed binding site, and the result with the longest alignment is used as the corresponding binding site. A transformation matrix is calculated to superimpose the moving binding site onto the fixed.
Source Code
Note that Nanome maintains a fork of the code from the original paper. The original codebase can be found here
Improvements to the codebase include:
- Updating to Python3
- Cut out unnecessary features for our use case
- Logging
Overview
References
Sankar S, Chandra N (2022) SiteMotif: A graph-based algorithm for deriving structural motifs in Protein Ligand binding sites. PLoS Comput Biol 18(2): e1009901. https://doi.org/10.1371/journal.pcbi.1009901
Le Guilloux, V., Schmidtke, P. & Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinformatics 10, 168 (2009). https://doi.org/10.1186/1471-2105-10-168