Appearance
Minimization
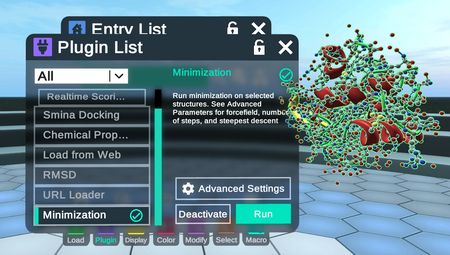
The minimization plugin optimizes the conformation of the selected molecule or complex to find a geometry where interatomic forces are as close to zero as possible. The molecular mechanics methods developed by OpenBabel are employed.
Instructions
To use the minimization plugin, Activate it from the plugin menu, and select one or more complexes from the entry menu. At this point, you can press Run to use the default settings.
The minimization window is also accessible in the Modify Menu under the Compute Tab. Click on the gear wheel icon to access settings.
Auto-minimize is a separate feature that can be toggled on or off in the Med Chem Tool under the Prep tab. Small molecules or edits to the protein will be minimized automatically when the feature is on. More information can be found in the Med Chem Tool documentation.
The minimization process can be stopped at any time by pressing the 'Stop' button.
Settings
Settings can be viewed and changed before running the minimization from the Advanced Settings icon button; this is the “gear wheel” in the main minimization window.
Force fields that are supported in Nanome: General AMBER, Ghemical, MMFF94, MMF94s, and Universal. The default force field is Universal.
Choose from two first derivative techniques:
Steepest Descent to find a local minimum, best suited for starting structures far from the minimum;
Conjugate Gradients, which accumulates stepwise information toward convergence and is best suited for larger systems.
Select up to 5000 steps (default is 2500) and press Run with the trigger button. The selected structures will have their bounding boxes turned on and their energies minimized.