Appearance
Chemical Interactions
The Chemical Interactions plugin visualizes ligand-receptor and protein-protein interactions. It utilizes arpeggio for calculating interactions.
Instructions
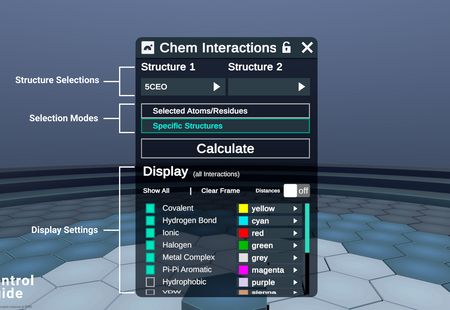
Select Structure 1 from the dropdown. This should be the receptor, or the larger of the two interacting proteins
Set your Selection mode for Structure 2
- Selected Atoms/Residues: Uses the atoms/residues selected in the workspace, and calculates interactions with Structure 1.
- Atoms/residues can be from any complex in the Entry List.
- Does not require the Structure 2 dropdown
- Specific Structures: Select either another complex in the workspace, or a ligand extracted from Structue 1.
- When Structure 1 dropdown is populated, the plugin automatically detects ligands, and adds them to the choices for Structure 2.
- For performance, you want to minimize the number of atoms in your selections for Structure 2. Doing a full protein-protein calculation can cause long render times, so we advise you narrow down your selection as much as possible.
- Selected Atoms/Residues: Uses the atoms/residues selected in the workspace, and calculates interactions with Structure 1.
Press Calculate, which will identify interactions using arpeggio, and render results in your workspace.
Modify the Display Settings to show or hide interactions of interest.
- Change the visibility or color of each interaction type
Hide All: Quickly Hide all interaction lines. Toggles through Hide All, Show All, and Show DefaultClear Framebutton will delete all interaction lines currently visible in the workspace.