


New Updates

New LiveDesign Plugin Documentation
Explore our new LiveDesign plugin that allows you to seamlessly load molecular data from LiveDesign and send ligand poses to LiveReports. Authenticate with tokens, filter entities, and visualize 3D data directly in Nanome.
Learn MoreVideos of Spatial Recordings
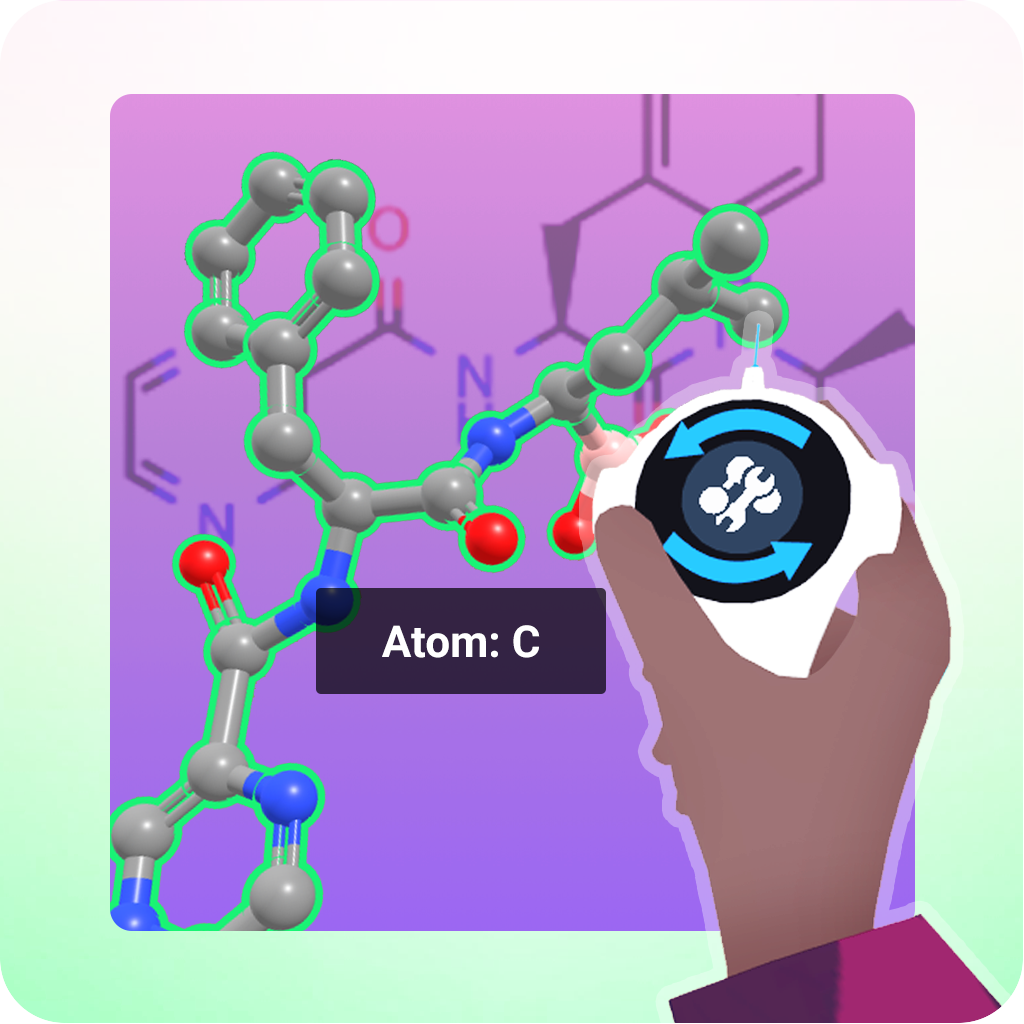
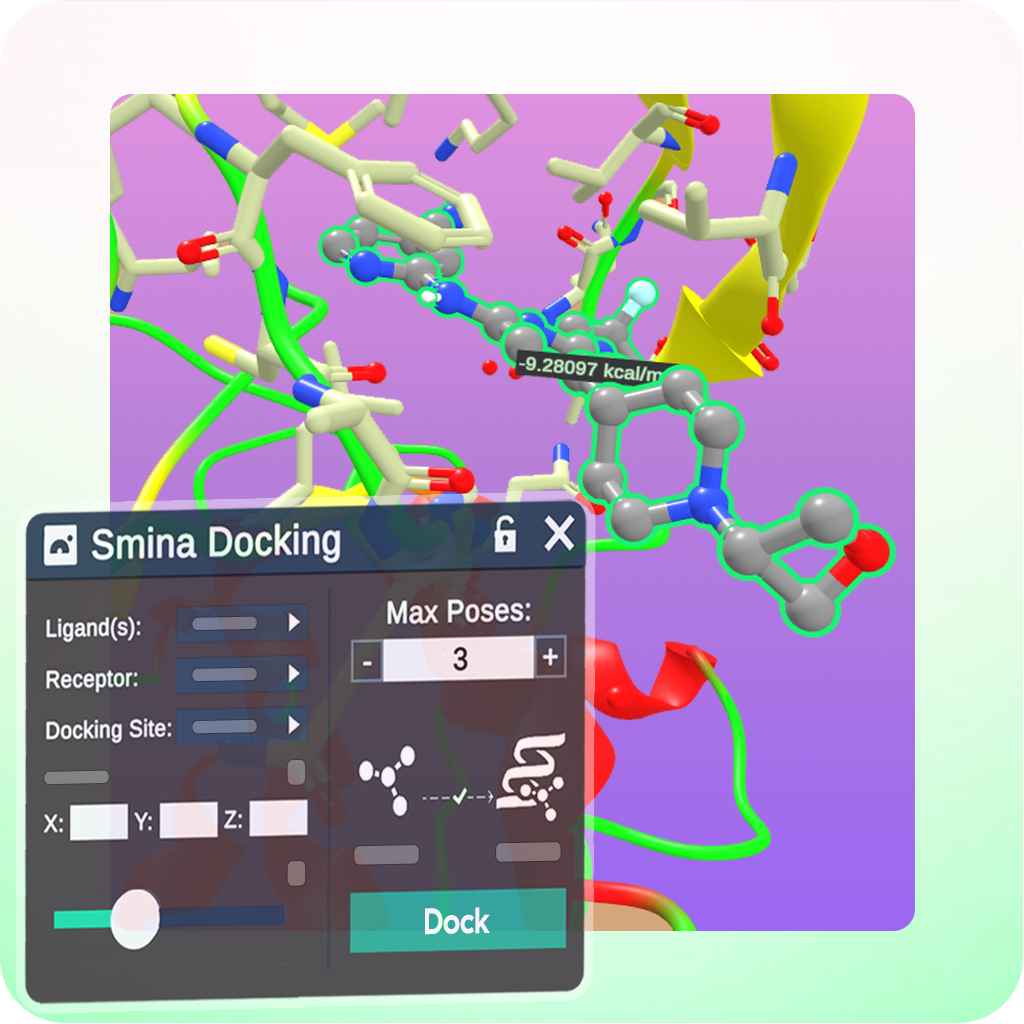


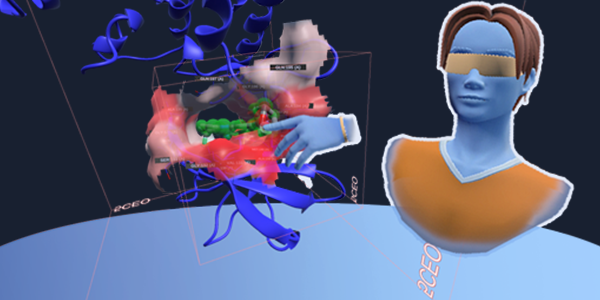

Advanced Tutorials
Nanome Youtube Channel









Nanome Blog

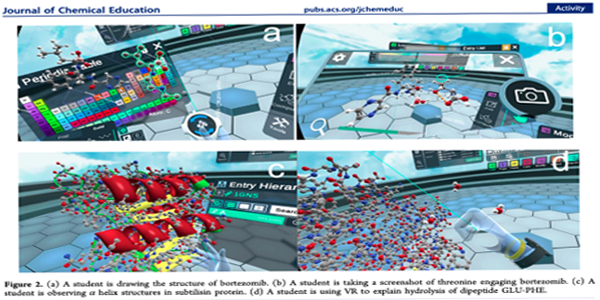


Featured Sections
Main Menus
All the menus of NanomeTools Menu
All the tools of NanomeNanome Stacks
An overview of the plugin systemCompatible Headsets

Requirements
Overview →